


Optogenetics could change the world. So why hasn’t it?
2.9k words, 14 minutes reading time

Note: This is a (my first ever!) guest post, written by Pelagia Martin, an undergraduate biology student at Stanford. It is over a question I myself have been wondering for a very long time, and it was great to read a post answering it. And a reminder: there’s an NYC bio-ML meetup happening on April 16, here’s a Partiful w/ more details.

Introduction
Optogenetics is a technique in bioengineering invented in the early 2000s where light can be used to have direct control over neuron activity. The central mechanism revolves around channelrhodopsins, ion channels and pumps found primarily in species of bacteria and algae, that open when triggered by light. Similar to human eyes where rhodopsin acts as a photoreceptor that converts light into chemical signals that communicate vision to the brain, channelrhodopsins used in optogenetics change conformation when exposed to light such that ions can enter the neuron and trigger neuronal excitation or inhibition.
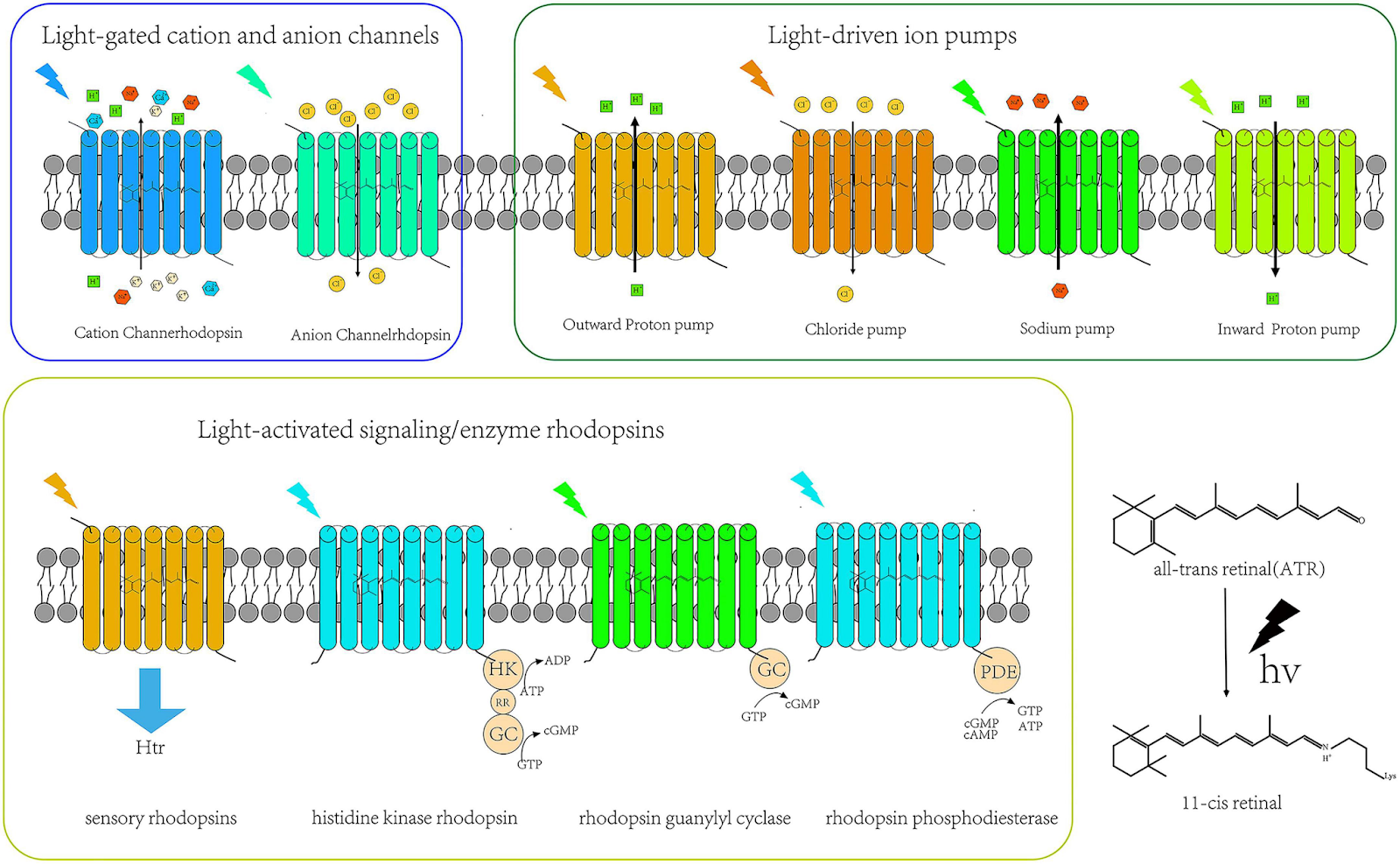
But if we’ve implied that rhodopsin (naturally expressed in retinal rod cells) does the same functional thing as channelrhodopsins (only made in bacteria), why use the latter? We’d need to do genetic engineering regardless, since rhodopsin only exists in one type of human cell, but it is a fair question. The answer lies in how these proteins respond to light and what they do downstream.
Rhodopsin, while light-sensitive, doesn’t directly alter ion flow. Instead, it activates a G-protein signaling cascade that eventually modulates ion channels indirectly. Helpful for complex mammalian vision applications, but too slow and complex for precise, moment-to-moment control of neuron firing.
Channelrhodopsins, on the other hand, are light-gated ion channels themselves. They cut out the middleman: when exposed to light, they change conformation and instantly open to allow ions to pass through, directly shifting the membrane potential of the neuron, forcing it to fire.
Thus, if we can force a neuron to produce channelrhodopsins, it gives scientists a button by which they can precisely turn it off and on, at the scale of femtoseconds. That is, as long as light can reach it — more on that later. But first: why do this at all?
Applications
This has a multitude of applications in basic neuroscience research such as loss and gain of function studies, as brain regions can be “shut off” for necessity studies without needing to cause brain lesions that cause permanent damage to the model. Likewise, optogenetics can be used for sufficiency studies by triggering excitation of neurons on demand and observing the effect without having to use electric stimulation. Invasiveness and risk of harm to model organisms is a big setback in many basic neuroscience experiments, so being able to minimize this with optogenetics is highly applicable. Additionally, with the ability to choose which neuronal cell type channelrhodopsins are expressed in, we can more accurately examine the role of different groups of neurons in various brain and body functions.
Optogenetics also has impressive potential for application in healthcare. One of the most widely discussed applications is in blindness caused by retinal neurodegeneration. A group led by José-Alain Sahel was actually able to partially restore vision in a patient with retinitis pigmentosa by injecting channelrhodopsin proteins into their affected eye and having the patient wear light-stimulating goggles. Before stimulation from the goggles the patient had no ability to perceive objects, but during stimulation could perceive, locate, and grab objects in front of them. This is incredibly exciting, and hopefully will become an approved treatment for blindness caused by neurodegenerative eye diseases.
Blindness is a great place to start with optogenetic treatments as there is already a system in place for light to reach neurons that express channelrhodopsins (via the eye). Things get slightly more complicated for other areas of the body, but nonetheless have their own exciting interventions being developed. One of my personal favorites is the application of optogenetics as a treatment for depression and depressive-state symptoms. For example, a group led by Allyson Friedman found that by hyper-stimulating midbrain dopamine neurons depression phenotype could be reversed. Moreover, optogenetic research is helping neuropharmacologists discern what reward pathways are impacted in depression for better development of antidepressive agents for treatment-resistant patients.
This list of applications is nowhere near exhaustive, and the potential of optogenetics as a technique is really exciting. You may be wondering, if this technique was invented in the early 2000s, why haven’t we heard more about it? Why haven’t we cured blindness, or depression, or any of the number of ailments that optogenetics could treat?
As is often the case with world-changing scientific technologies, it is not without limitations. In vitro, optogenetics is not that difficult. Cells can be transfected to take up your channelrhodopsin-encoding DNA (typically with electroporation, but you could use a viral vector or CRISPR if you were feeling fancy), expression levels can be checked easily since channelrhodopsins are fluorescent, and light can be shone on them without barriers that block electromagnetic waves or alter the wavelengths of waves that pass through. Easy peasy lemon squeezy.
The roadblocks
All of this changes when we go in vivo. Crucial to conducting in vivo optogenetic manipulation are two particularly foreboding hurdles: gene therapy and getting a light source inside the body. Gene therapy and the light source don’t only cause issues upon initial insertion, but require long term maintenance that can be costly and invasive. Any method that relies on gene therapy is bound to encounter some pretty tricky issues. Gene therapy was first successfully used in 1990, but there were no FDA approved treatments until 2017, nearly 30 years later. The primary issues keeping gene therapy from widespread use are less about how difficult the method is, and more about how difficult it is to mass-produce. Gene therapy is expensive and individualized by nature, so it is largely not worth it for pharmaceutical companies to put in the effort to develop treatments. Additionally, gene therapy isn’t without risk.
Gene therapy is hard
The primary method used in gene therapy for introducing new genetic information is with viral vectors. Lentiviruses are commonly used as these vectors, but can be genotoxic and potentially cause cancer. Additionally, lentiviral gene insertion is essentially random, and if a promoter were to be inserted near an oncogene it could also cause cancer. Site-specific gene editing like CRISPR comes with the risk of off-site deletions that, you guessed it, can cause cancer (and lots of other bad things that lead to death and disability). Lentiviruses are starting to fall out of style in favor of adeno-associated virus (AAV) vectors (Dyno Therapeutics plug), a generally safer alternative to lentiviruses. The major setback of AAVs is combatting the immune system so genetic material can be inserted. Major immune reactions aren’t incredibly common, but when they happen they can be deadly.
The light source problem
Say the complications of gene therapy are all solved. We are still left with an incredibly daunting engineering challenge of getting light inside the body. With the earlier case of blindness, goggles were designed that allowed light to reach retinal cells with minimal invasiveness. This is not possible anywhere else in the body, and much more invasive methods must be used. In mouse models, you can pretty easily insert optical fibers attached to an LED over a hole in their skull, as shown.
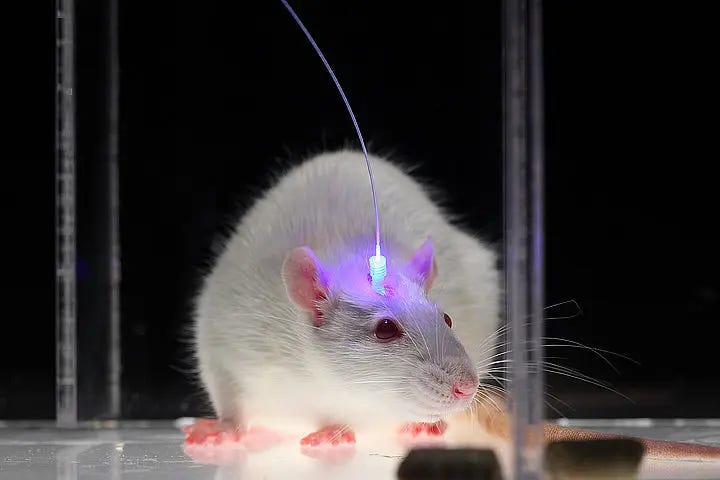
One can imagine it's pretty unlikely people are going to be walking around wearing giant LED hats with holes in their skulls. This is incredibly invasive, and less than fashionable. Different intervention locations come with different complications. For example, if your target neurons are in a sensitive tissue like neurons in the heart, optical fibers can irritate tissues and cause cardiac irregularities. In addition to the issue of invasiveness, it is difficult to keep these devices powered. Similar to cardiac pacemakers, light-emitting devices are typically charged with a battery. When the battery dies, the whole device has to be replaced, requiring yet another invasive surgery. The LED devices also heat up when they are continuously activated, which can be harmful to brain tissue.
There are also a number of difficult physics kinks yet to be worked out. When light passes through any non-air medium there is scattering that changes its path and lowers its wavelength. This optical scattering causes some serious distortion when light passes through brain tissue. This is a big issue when channelrhodopsins require precise neuron targeting and a specific wavelength of light in order to be activated. In order to compensate for this effect you’d have to send more intense light into the brain, at a higher wavelength than what you would eventually need to stimulate your channelrhodopsin. This, in turn, changes for each new brain area due to heterogeneity in tissue depth and density. Additionally, the more intense light you send into the brain, the more you risk brain tissue heating and causing permanent damage.
Maintenance
Compounding all of this, optogenetic therapy is a long term therapy with a long term patient commitment. All of the components of the therapy need pretty frequent maintenance, as channelrhodopsin expression can decrease over time and light-producing devices can have mechanical issues, in addition to the aforementioned issue of needing to be charged. Maintaining expression of a genetic intervention is a common problem for gene therapy. If the gene is recognized as foreign after being integrated into the DNA the whole cell could be killed by the immune system. If the immune system is successfully suppressed and the DNA is integrated without any problem, genes introduced via gene therapy and their promoters are often silenced by post-translational modifications like DNA methylation. It might be worth exploring inserting an extra copy of a gene that is frequently expressed in the neuron along with our target gene to see if this silencing can be reduced. For example, a neurotransmitter release-associated gene. If we could link the expression of the neurotransmitter release gene to our channelrhodopsin such that both genes are undergoing transcription often, it is less likely that our channelrhodopsin would be silenced by post-translational modification (this is of course just speculative, but I would love to see someone try it).
As of now though, gene therapy requires re-dosing to ensure channelrhodopsins stay at therapeutic levels. This means more risk of life-threatening immune reactions and painful injections into sensitive areas. To repair or charge light-producing devices they must be surgically removed, raising the risk of infections and the many other complications associated with surgery.
If a patient decides all of these issues aren’t worth the benefit or can’t afford the maintenance, it’s not an easy thing to stop. All gene therapies are meant to be permanent, and channelrhodopsins will continue to be expressed in your neurons whether you like it or not (though they may be present below therapeutic levels). If your brain had adapted to the stimulation of its neurons, conditions that were being treated may revert back to even worse symptoms than patients started with. For example, if you were treated for depression with an optogenetic intervention where neurons were stimulated to release more serotonin, if those neurons are suddenly no longer stimulated there would be an extreme drop in serotonin levels. These neurons may have previously been releasing low yet present levels of serotonin that caused your depression phenotype, but now may release none at all because their threshold for sending the serotonin release signal has changed. This is incredibly dangerous, and it is important we have systems in place to support long-term patient care before widely implementing these therapies.
How do we fix it?
Though difficult to overcome, none of these issues are total dealbreakers in my opinion. Luckily, there’s lots of research in progress to fill in the gaps to making optogenetics truly great. In the gene therapy arena, we’re figuring out how to bypass the immune response triggered by AAV vectors by using similar techniques to organ transplants. In addition to using standard immunosuppressants, researchers are using plasmapheresis, where plasma is removed from the body and all anti-AAV antibodies are lost with it. This eliminates basically all immune response, allowing AAV vector DNA to successfully enter the host genome while minimizing harm. Additionally, AAV vectors engineered to have fewer CpG motifs (oligonucleotides found primarily in pathogens) have been found to further minimize immune response. All of these methods combined could make using AAV vectors nearly harmless, and could revolutionize optogenetics and the larger field of gene therapy.
On the light-emitting device side of things, devices are being designed that keep batteries and other mechanical components that may need repair outside of the body while optic fibers transmit light to the target neurons. MicroLED probes have also been created that can be implemented with minimal invasion, reducing the risks associated with surgery to implant larger light sources. These flexible, silicon-based arrays can be implemented just under the skull, greatly reducing the risk of damage to brain tissues, making them a preferable intervention compared to optic fibers for cortex stimulation. MicroLED probes also allow for reduced optical scattering due to their spatial specificity and small distance from the light source to the target tissue. These probes still cause brain tissue to heat up, but at a slower rate than a larger LED or directed laser.
There are also many developments happening in the engineering of channelrhodopsins themselves. As more channelrhodopsins are discovered, researchers are beginning to figure out which are maximally effective for different purposes. For example, channelrhodopsins that absorb red light can be used in deeper areas of the brain, as red and near-infrared light can travel deeper into the brain with less optical scattering. Studies using machine learning are being conducted to design channelrhodopsins that require less light to produce neuronal excitation, so signals can be sent while minimizing the heating of brain tissue. Additionally, structural studies are being done that can elucidate what parts of channelrhodopsin structure are most important for ion conduction, and how we can improve the channels that we have to be faster and more efficient.
The only roadblock to optogenetics being used widely that could really delay progress is the economic problem of gene therapy. Hopefully with the development of more AI tools for biology, we will be able to optimize gene therapy to a point that its production cost is low enough for us to see a major surge in commercially available therapies.
Is it worth it?
It is worth considering that all of these setbacks might make optogenetics not worth it. After all, there is more research right now in how to fix optogenetics than how to apply it clinically.
That said, I think optogenetics has some key advantages over other neuromodulation techniques that make it worth it to work the kinks out. These other techniques, chemogenetics and magnetogenetics, open ion channels via small molecule binding to designer receptors and magnetic field-generated torques mechanically opening ferritin-bound ion channels, respectively. Compared to optogenetics both techniques lack temporal and spatial resolution, and could have unknown off-target effects that light does not.
Ligands used in chemogenetics could potentially have unknown impacts on other cell receptors and tissues despite being designed to only bind with designer receptors. Additionally, chemical binding takes much more time to turn on or off compared to the femtosecond scale optogenetics operates on. Magnetogenetics is much faster than chemogenetics, but still doesn’t quite reach the same temporal resolution as optogenetics. It can also disrupt electronic devices in the body like pacemakers if the magnetic field is strong enough, as well as potentially inducing unwanted voltages in cells other than the target, although the extent at which these unwanted voltages cause any harmful effect is debatable.
Finally, timing is of the essence in many basic neuroscience studies and clinical applications. If you wanted to use any of these techniques to treat something like cardiac arrhythmia, you would want to inhibit the abnormal heart rhythm as quickly as possible. However, both of these techniques come with the incredible benefit of being almost entirely surgically noninvasive.
But, the beauty of all of these methods is that none of them exist alone, and the three could be used in different situations where their unique benefits are best suited. For example, optogenetics could be used to reverse neurodegenerative blindness, as light can easily enter the eye so invasive devices wouldn’t be used and a quick temporal response is necessary. Chemogenetics or magnetogenetics could be used in depression treatment, where quick temporal response isn’t as important. I hope to see further development of chemo- and magneto- genetics in addition to optogenetics to fill out our bioengineering toolbox, in order to fully explore the possibilities of neuromodulation.
Overall, I think optogenetics is an important technique that will see widespread clinical applications, especially in the areas where it can be applied least invasively. In combination with other bioengineering techniques, I believe that optogenetics could certainly change the world. It’s just going to take some time.
 | A guest post by
|